SPECIALIZED PRIMARY CARE SERVICES FOR ADULTS WITH SICKLE CELL DISEASE
TAIBU has established the first specialized primary care services for adults with Sickle Cell disease in the GTA. If you are an adult living with sickle cell disease or a care giver, you can access our clinic on
Tuesdays from 1:30 pm – 7:30 pm.
Evaluation of our primary care service has demonstrated that adults who have attended the clinic have significantly reduced the number of Emergency Department visits as well as their hospitalization.
Clients have also been able to increase their knowledge of the unique impact of their chronic condition and how best to manage it.
If you are an adult with sickle cell disease please contact us for access: (647) 644-3936
MONTHLY SUPPORT GROUP
Members of the sickle cell community have initiated a self help group that meets on a monthly basis to support each other, increase their knowledge and capacity by inviting guest speakers to come and talk to them, to have gentle exercise and relaxation classes, to advocate for each other and to have fun.
The Group meets every third Saturday of the month at TAIBU Community Health Centre.
For more information please call (416) 644-3539 (Ext. 2226)
ABOUT SICKLE CELL DISEASE
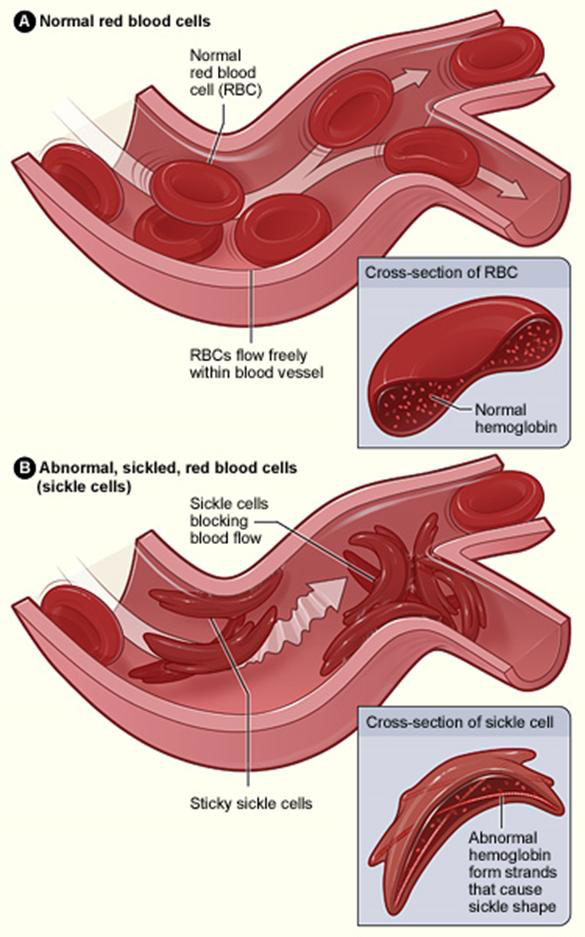
Sickle cell disease is an inherited disorder that affects hemoglobin, the molecule in red blood cells that delivers oxygen to cells throughout the body. People with this disorder produce an abnormal ‘hemoglobin’ called hemoglobin S, which can distort red blood cells into a rigid sickle, or crescent, shape.
Signs and symptoms of sickle cell disease usually begin in early childhood. Characteristic features of this disorder include a low haemoglobin level (anemia), repeated infections, and periodic episodes of pain. The severity of symptoms varies from person to person. Some people have mild symptoms while others are frequently hospitalized for more serious complications.
The signs and symptoms of sickle cell disease are caused by the sickling of red blood cells. When red blood cells sickle, they break down prematurely, which can lead to anemia. Anemia can cause shortness of breath, fatigue, and delayed growth and development in children. The rapid breakdown of red blood cells may also cause yellowing of the eyes and skin (jaundice). Painful episodes can occur when sickled red blood cells, which are stiff and inflexible, get stuck in small blood vessels. This deprives tissues and organs of oxygen-rich blood and can lead to organ damage, especially in the bones, lungs, kidneys, spleen, and brain. A particularly serious complication of sickle cell disease is high blood pressure in the blood vessels that supply the lungs (pulmonary hypertension). Pulmonary hypertension occurs in about one-third of adults with sickle cell disease and can lead to heart failure.
A person receives the sickle cell genes at the time of conception and they are born with them. Neither sickle cell trait nor sickle cell disease can be acquired later on in life. By the same token, people cannot lose their sickle cell genes over time. A person born with Sickle Cell trait (one sickle cell gene) will always have sickle cell trait. The same is true of sickle cell disease (two sickle cell genes).
People who inhert two genes for hemoglobin S (one for each parent) have sickle cell disease.
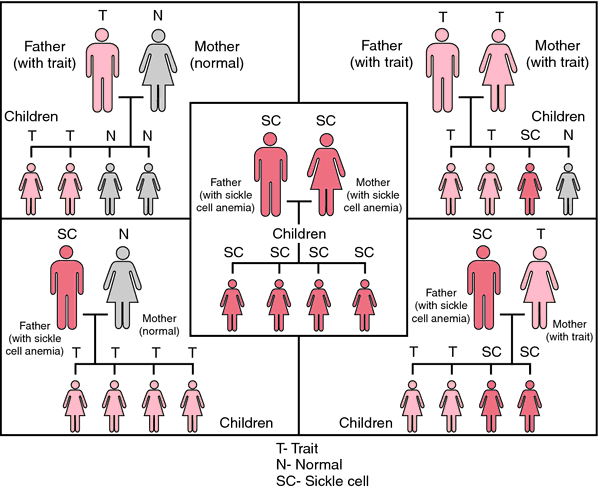
A child can inherit sickle cell disease only if both parents have sickle cell trait. The illustration below shows the possible combination of genes that can occur when one or both parents have sickle cell trait or related disorders.
If both parents have sickle cell trait, there is one in four (25%) chance that a child will inherit two normal genes from the parents. A one in four chance also exists that a child will inherit two sickle cell genes and have develop sickle cell disease, while in two chance exists that the child will inherit a normal gene from one parent and a sickle cell gene from the other. This would produce sickle cell trait.
If only parent has the trait there is a 50% chance that each child will be normal and a 50% chance that each child will have sickle cell trait. There is no chance that a child with sickle cell disease will be produced in this scenario.
Hemoglobin consists of four protein subunits, two subunits called alpha-globin and two subunits called beta-globin. The HBB gene provides instructions for making beta-globin. A particular mutation in this gene produces an abnormal version of beta-globin known as hemoglobin S (HbS). Other mutations in the HBB gene can occur. One of these causes the production of hemoglobin C (HbC) another disorder seen primarily in the population from Africa. HBB gene mutations can also result in an unusually low level of beta-globin; this abnormality is called beta thalassemia.
In people with sickle cell disease, both HBB genes (one from each parent) are abnormal so only hemoglobin S can be produced. . In sickle cell trait, one HBB gene is normal and one is not: these patients therefore produce a mixture of normal haemoglobin (A) and HbS. In other types of sickle cell disorder, one HBB gene is abnormal, producing hemoglobin S, while the other produces a different abnormal variant, such as hemoglobin C. For example, people with sickle cell-hemoglobin C (HbSC) disease have both hemoglobin S and hemoglobin C in their red blood cells. If mutations that produce hemoglobin S and beta thalassemia occur together, individuals have hemoglobin S-beta thalassemia (HbSBetaThal).
Sickle cell disease affects millions of people worldwide. It is most common among people whose ancestors come from Africa, Mediterranean countries such as Greece, Turkey and Italy; the Arabian Peninsula; India and Spanish speaking regions in South America, Central America and parts of the Caribbean.
Sickle cell disease is the most common inherited blood disorder in the United States, affecting 70,000 to 80,000 Americans. The disease is estimated to occur in 1 in 500 African Americans and 1 in 1,400 Hispanic Americans.
We don’t have an accurate estimate of people affected by sickle cell disease in Canada. However, it is reasonable to assume there would be 700-1000 patients who live with sickle cell disease in the Greater Toronto Area.
It is crucial that we all know our sickle cell status so that we are able to make informed decisions in our lives and the lives of our families.
TAIBU CHC organizes community awareness and screening events in the community. If organizations would like to invite TAIBU CHC to their events for Sickle Cell awareness and screening events please contact us at (416) 644-3539 (Ext. 226)
Sickle cell disease is a preventable disease.
The first step towards prevention is to know your sickle cell status. A simple blood test can determine if you have sickle cell trait or the disease. Everyone from the population at risk should have this test done even if they have no symptoms or signs to suggest a sickle cell disorder: people will the trait will usually not have any symptoms.
Good nutrition, while essential for anyone, is critical for patients with sickle cell disease. Some dietary recommendations include:
Fluids are number one in importance. The patient should drink as much water as possible each day to prevent dehydration.
Diet should provide adequate calories, protein, fats, and vitamins (especially folic acid and vitamin D) and minerals (especially iron and calcium). Patients and families should discuss vitamin and mineral supplements with their doctors and nurses.
Some studies claim that omega-three fatty acids, found in fish and soybean oil as well as dietary supplements, might make red blood cell membranes less fragile and possibly less likely to sickle, although no studies have proven this definitively.





